TOML files¶
Readfish experiments are configured using TOML files, which are minimal and easy-to-read markup files. Our configuration only uses tables and arrays of tables populated with key-value pairs. The TOML file contains almost all the information required for running readfish, such as information on barcodes, control/analysis regions, what basecaller or aligner to use. There are several example TOMLS, with comments explaining what each field does, as well as the overall purpose of the TOML file here - https://github.com//LooseLab/readfish/tree/main/docs/_static/example_tomls.
Data model¶
To understand each section of the TOML file it helps to understand the work that readfish does:
Initialise a connection to the MinKNOW API via the Read Until API
Initialise a basecaller
Initialise an aligner with a pre-computed reference file
Begin streaming live data from the live sequencing experiment
For each batch of the Read Until API read chunks:
Basecall the raw signal chunks
Align the basecalled chunks
Parse the alignments using the experiment configuration, determining whether the read is on or off target, then pass the decision to the Read Until API for it to be effected on the sequencer
(Optionally) Check for updated configuration parameters with a new reference or targets
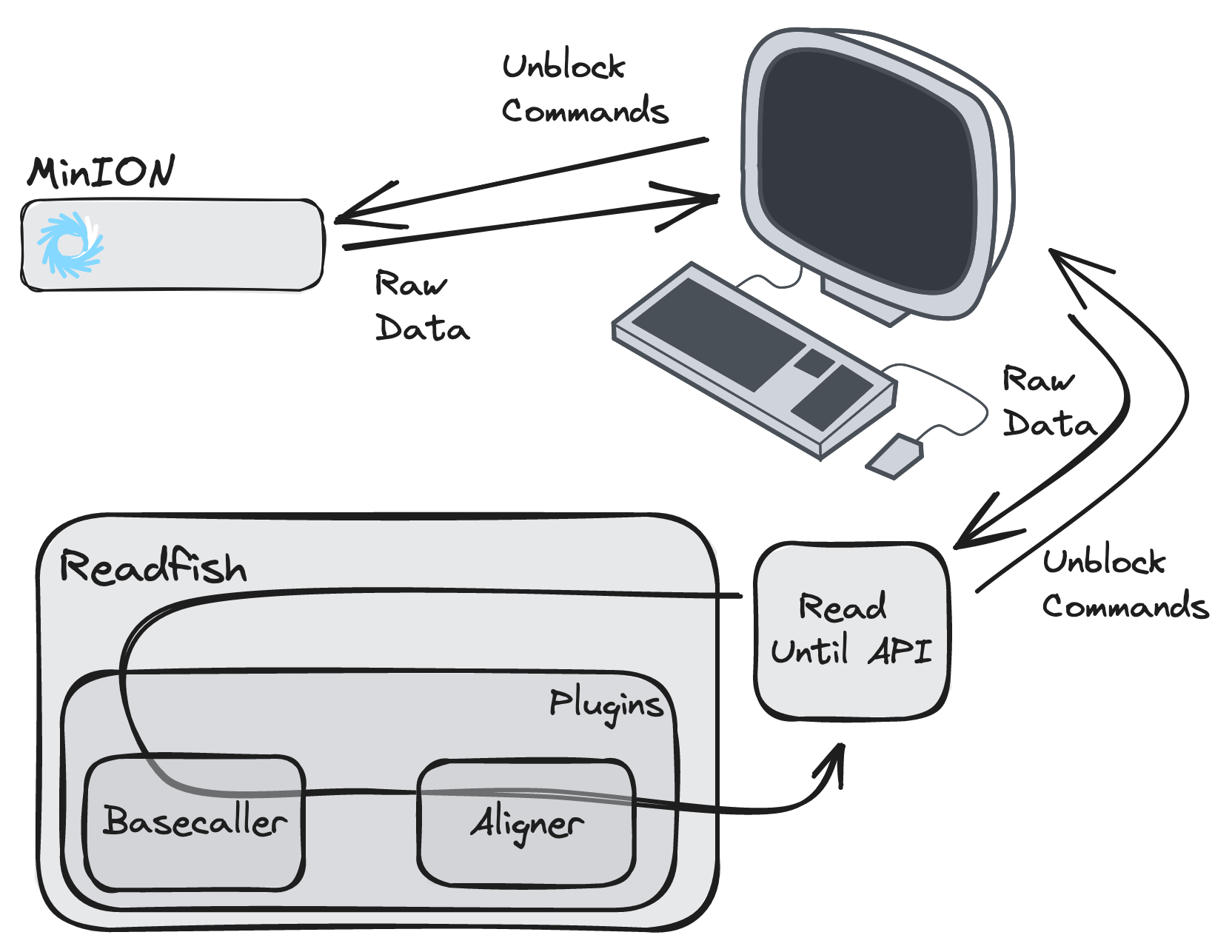
Flow diagram of data flow from the sequencer to readfish and back.¶
As readfish is designed around the need to both basecall and align data we have two TOML sections for configuring these tools.
The first, caller_settings, controls the basecaller and the second, mapper_settings controls the aligner.
These are expanded on in the plugin configuration section.
In addition readfish also manages barcodes and analysis regions on the flow cell. These allow for arbitrary configuration schemas that match your experimental needs. These are expanded on in the barcode configuration and analysis regions sections.
Plugin configuration¶
Readfish uses “plugins” to configure basecalling and alignment.
We provide some default plugins using Dorado (via ont-pybasecall-client-lib) and mappy (via mappy), for basecalling and alignment respectively.
The way that you tell readfish what plugin to load is by specifying it after the table name, for example:
[caller_settings.dorado]
# ^^^^^^
# Chooses the Dorado plugin
This indicates that readfish should use Dorado for basecalling.
Basecaller¶
Dorado¶
This section is specific to the Dorado plugin for basecalling.
The caller_settings.dorado [table] specifies the basecalling parameters used by the basecaller.
An example table is given below:
[caller_settings.dorado]
config = "dna_r10.4.1_e8.2_400bps_hac"
address = "ipc:///tmp/.guppy/5555"
debug_log = "basecalled_chunks.fq" #optional
The only required keys are address and config these indicate to Dorado what server to connect to and what basecalling model to load.
The config parameter must be a valid Dorado configuration excluding the file extension; these are found in the Dorado data folder (on linux ls /opt/ont/dorado/data/*.cfg).
The address is the path to the IPC socket Dorado is listening on, for Dorado servers run by MinKNOW the default is ipc:///tmp/.guppy/5555. Why guppy? Blame history…
The debug_log is an optional file that the basecalled FASTQ are written to.
Key |
Type |
Description |
Required |
|---|---|---|---|
|
string |
Name of base calling config |
True |
|
string |
Address of Dorado socket - default ipc:///tmp/.guppy/5555 |
True |
|
string |
Optional - Filepath to write out base-calls to - should end in a FASTQ suffix. |
False |
Any extra key value pairs are passed verbatim to the PyBasecallClient instance upon initialisation as keyword arguments.
For example:
server_file_load_timeout = 60
Will set the server_file_load_timeout parameter to 60 seconds for Dorado.
Example Dorado server parameters
- PyBasecallClient.set_params(self: pybasecall_client_lib.client_lib.BasecallClient, params: dict) apps::Result[source]
Parameter setter for connection to the server.
- Parameters:
params (dict) – Dictionary of named parameters.
- Returns:
A code indicating whether the call was success.
The allowed parameters are:
client_name (str): A name for the client, which will appear in server log entries.
query_timeout (int): Milliseconds to wait for a server response before timing out. Default is 2000ms.
server_file_load_timeout (int): Seconds to wait for files to be loaded on the server. Default is 180s.
- pass_read_timeout (int): Default value to use if the optional pass_read parameter wait_duration_ms is
not supplied. The default value for this is zero, which if applied means pass_read will be non-blocking.
connection_timeout (int): Milliseconds to wait for a server connection attempt. Default is 15000ms.
- reconnect_timeout (int): Seconds to wait for the client to reconnect to the server if the connection is
broken. Default is 300s.
max_message_size (int): Size of blocks to be sent to the server, in samples. Default is 256000.
max_reads_queued (int): Maximum number of reads (or read batches) to queue for sending to the server. Default is 20.
- max_server_read_failures (int): Maximum number of times a read can be in-flight when the client has to reconnect before the
client will stop resubmitting the in-flight reads. Default is 10.
priority (ReadPriority): Priority of the client (low, medium, or high). Default is medium.
move_enabled (bool): Flag indicating whether to return move data. Default is False.
barcode_kits (list): Strings naming each barcode kit to use. Default is to not do barcoding.
require_barcodes_both_ends (bool): Flag indicating that barcodes must be at both ends. Default is False.
enable_trim_barcodes (bool): Option indicating that barcodes should be trimmed. Default is False, set to true to enable trimming.
trim_adapters (bool): Flag indicating that adapters should be trimmed. Default is False.
trim_primers (bool): Flag indicating that primers should be trimmed. Default is False.
align_ref (str): Filename of index file to use for alignment (if any). Default is to not align.
bed_file (str): Filename of BED file to use for alignment (if any). Default is to not align.
barcode_list (list): An optional list of barcodes to filter against. Default is no restriction.
pair_by_channel (bool): An optional flag to enable caching by channel for duplex pairing. Default is False (caching by time).
- chunk_size (int): For adaptive sampling. Specify the chunk-size for basecalling. If you are truncating reads, send the
value you are truncating to.
Possible return values are:
success: The parameters have been set.
already_connected: The parameters cannot be changed after the client is connected to the server.
failed: The parameter dict was not formatted correctly.
To see parameters that your Dorado installation has run the following python snippet in your readfish environment
PAGER=cat python -c "import pybasecall_client_lib.pyclient as pgc; help(pgc.PyBasecallClient.set_params)"
Basecaller no operation (no_op)¶
In addition to the Dorado basecaller we provide a “no operation” basecaller. This plugin does nothing. It will only iterate the live chunks of data and pass through the minimal amount of data that is needed for the next steps. This is added using:
[caller_settings.no_op]
Aligner¶
minimap2¶
Currently we provide two mapping plugins, mappy, and mappy-rs.
Happily these both take the same fields in the TOML file, so you can switch between them without changing your TOML file parameters.
Currently, these plugins take the same TOML file plugins section, and which Aligner is used is chosen based on the TOML mapper_settings section name.
.. warning::
It is worth noting that for PromethION runs, mappy will not be able to keep up and mappy-rs with at least four threads is recommended.
For mappy (single threaded)
[mapper_settings.mappy]
# ^^^^^^
# Chooses the mappy plugin
For mappy-rs (multi threaded)
[mapper_settings.mappy_rs]
# ^^^^^^
# Chooses the mappy-rs plugin
The fn_idx_in field is the path to the reference, accepting either a precomputed minimap2 index, an .mmi file, or a uncompressed or gzipped fasta file.
We strongly prefer an already created MMI.
The debug_log field is the file name that the mappings produced by mappy-rs/mappy are written into, in the PAF format.
mappy-rs also takes an optional n_threads field, which determines the number of threads which are used for alignment.
n_threads defaults to 1, so if you need more you must set it!
[mapper_settings.mappy_rs]
fn_idx_in = "/Path/To/Reference.mmi"
n_threads = 4
debug_log = "mapped_chunks.paf"
Key |
Type |
Description |
Required |
|---|---|---|---|
|
string |
Absolute path to the reference FASTA/mmi |
True |
|
string |
File to write PAF records to from alignments seen |
False |
|
int |
Optional - if using |
False |
Any extra key value pairs are passed varbatim to the Aligner instance upon initialisation as keyword arguments.
For example:
best_n = 1
Will set the best_n parameter on the Aligner instance to 1.
All available keyword arguments can be seen on the mappy documentation
Aligner no operation (no_op)¶
In addition to the minimap2 based aligners we provide a “no operation” aligner.
This plugin does nothing.
It will only iterate the Results from the basecaller and pass them through to the next steps.
This is added using:
[mapper_settings.no_op]
Analysis regions¶
Analysis regions are the arrays of tables that define different experimental conditions over the surface of the flow cell. For experiments that do not use barcodes you must have at least one analysis region.
The physical layout of each flow cell constrains how many experimental conditions can be used as the number of channels in each region must be equal. The number of regions subtables determines how many times the flow cell is divided. The maximum number of regions for MinION flow cells is 32 and for PromethION flow cells is 120. The number of conditions must be a factor of the number for the selected combination.
The flongle is a strange shape - 13 columns by 10 rows. 13 is a prime, meaning it cannot be split vertically (except into 13), so flongles must be split horizontally into 2,5 or 10 regions.
As an example applying two analysis regions to the layout below would split the flow cell into left/right regions.
+------------+ +------+------+
| 1 2 3 4| | 1 2| 3 4|
| 5 6 7 8| --> | 5 6| 7 8|
| 9 10 11 12| | 9 10| 11 12|
+------------+ +------+------+
The flow cell can be split both vertically and horizontally, with the split_axis parameter in the TOML file deciding this.
The axis can be either 0 or 1, with 0 splitting horizontally and 1 splitting vertically.
The default value is 1, so this only needs setting if you wish to split horizontally.
Vertical (axis=1) Horizontal (axis=0)
+------------+ +------+------+ | +------------+ +------------+
| 1 2 3 4| | 1 2| 3 4| | | 1 2 3 4| | 1 2 3 4|
| 5 6 7 8| --> | 5 6| 7 8| | | 5 6 7 8| --> | 5 6 7 8|
| 9 10 11 12| | 9 10| 11 12| | | 9 10 11 12| +------------+
| 13 14 15 16| | 13 14| 15 16| | | 13 14 15 16| | 9 10 11 12|
+------------+ +------+------+ | +------------+ | 13 14 15 16|
+------------+
This set at the very top of the TOML file, like the channels parameter
split_axis=0
[caller_settings.dorado]
config = "dna_r10.4.1_e8.2_400bps_hac"
address = "ipc:///tmp/.guppy/5555"
debug_log = "basecalled_chunks.fq" #optional
......... # and so on
Experiments with one region¶
When wanting to apply a single targeting strategy over the flow cell a single region can be provided. This can be specified like so:
[[regions]]
name = "condition name"
min_chunks = 1
max_chunks = 4
targets = "path/to/targets.bed"
single_on = "stop_receiving"
multi_on = "stop_receiving"
single_off = "unblock"
multi_off = "unblock"
no_seq = "proceed"
no_map = "proceed"
A full table of keys that can be given can be found in region sub-table.
Experiments with more than region¶
Experiments with multiple regions (for example those with an analysis and control condition) regions can be specified for each condition. For example:
[[regions]]
name = "analysis"
control = false
...
[[regions]]
name = "control"
control = true
...
Regions sub-tables¶
Each Regions sub-table must contain all of the required keys, these are the same between barcoded and non-barcoded toml files.
Key |
Type |
Values |
Description |
Required |
|---|---|---|---|---|
|
string |
N/A |
The name given to this condition |
True |
|
bool |
N/A |
Is this a control condition. If |
False |
|
int |
N/A |
The minimum number of read chunks to evaluate |
True |
|
int |
N/A |
The maximum number of read chunks to evaluate |
True |
|
string or array |
N/A |
The genomic targets to accept or reject; see target formats |
True |
|
string |
[unblock, stop_receiving, proceed] |
The action to take when a read has a single on-target mapping |
True |
|
string |
[unblock, stop_receiving, proceed] |
The action to take when a read has multiple mappings with at least one on-target |
True |
|
string |
[unblock, stop_receiving, proceed] |
The action to take when a read has a single off-target mapping |
True |
|
string |
[unblock, stop_receiving, proceed] |
The action to take when a read has multiple mappings that are all off-target |
True |
|
string |
[unblock, stop_receiving, proceed] |
The action to take when a read does not basecall |
True |
|
string |
[unblock, stop_receiving, proceed] |
The action to take when a read does not map to your reference |
True |
|
string |
[unblock, stop_receiving, proceed] |
Optional. The action to take when a read has not been seen at least |
False |
|
string |
[unblock, stop_receiving, proceed] |
Optional. The action to take when a read has been seen more than |
False |
The meanings of unblock, stop_receiving, and proceed are given below.
proceed: Allow one more chunk to be captured, before trying to make another decision, i.e proceed for now
unblock: Unblock the read
stop_receiving: Allow the read to be sequenced, and stop receiving signal chunks for it.
Target Formats¶
The targets field on a region can accept either a string or an array of strings.
If a string is provided this should be a fully qualified path to a text file that consists of genomic targets in either as CSV or BED entries.
When an array is given all the elements in the array must conform with the non-BED formats.
Targets will be collapsed where they overlap.
BED or CSV targets¶
If the targets entry is a path to a file it must either be a six-column-BED formatted file or a CSV.
If a CSV file there should be no header and the coordinates should be given as
contig,start,stop,strand
As a concrete example:
chr1,1000,2000,+
chr2,3000,4000,-
Targets given in this format will only select (for or against) reads where the alignment start position is within the region on the given strand.
TOML array targets¶
If specifying targets in a TOML array they can be any of contigs or coordinates:
For example contigs:
>chr1 Human chromosome 1becomeschr1
Targets given in this format specify the entire contig as a target to select for or against.
Barcode specific configuration¶
If you are using Dorado for basecalling then the additional barcode_kits parameter is required on the caller_settings table.
[caller_settings.dorado]
config = "dna_r10.4.1_e8.2_400bps_fast"
address = "ipc:///tmp/.guppy/5555"
debug_log = "basecalled_chunks.fq"
barcode_kits = "EXP-NBD196"
Note, to specify multiple barcode kits, the format to use is a space separated string, not an array. For example, "KIT1 KIT2".
[[regions]] are not needed when using a barcoded schema.
Instead a TOML table named barcodes is required.
This table has two required sub-tables barcodes.classified and barcodes.unclassified.
These are the fallback configurations for unspecified barcodes.
For specific barcodes a full configuration table should be specified using the barcode name, e.g. barcodes.barcode01 for “barcode01”.
[barcodes.classified]
name = "classified_reads"
control = false
min_chunks = 0
max_chunks = 2
targets = []
single_on = "unblock"
multi_on = "unblock"
single_off = "unblock"
multi_off = "unblock"
no_seq = "proceed"
no_map = "proceed"
[barcodes.unclassified]
name = "unclassified_reads"
control = false
min_chunks = 0
max_chunks = 2
targets = []
single_on = "unblock"
multi_on = "unblock"
single_off = "unblock"
multi_off = "unblock"
no_seq = "proceed"
no_map = "proceed"
[barcodes.barcode01]
name = "barcode01_targets"
control = false
min_chunks = 0
max_chunks = 2
targets = ["chr1", "chr2"]
single_on = "stop_receiving"
multi_on = "stop_receiving"
single_off = "unblock"
multi_off = "unblock"
no_seq = "proceed"
no_map = "proceed"
Validating a TOML¶
We provide an entry point to validate TOML files - readfish validate
readfish validate experiment_conf.toml
Any errors that occur while loading the configuration will be written to the terminal.
As an example - if the reference is missing.
2023-06-27 16:07:03,041 readfish /home/adoni5/mambaforge/envs/readfish_dev/bin/readfish validate docs/_static/example_tomls/human_chr_selection.toml
2023-06-27 16:07:03,041 readfish check_plugins=True
2023-06-27 16:07:03,041 readfish command='validate'
2023-06-27 16:07:03,042 readfish log_file=None
2023-06-27 16:07:03,042 readfish log_format='%(asctime)s %(name)s %(message)s'
2023-06-27 16:07:03,042 readfish log_level='info'
2023-06-27 16:07:03,042 readfish prom=False
2023-06-27 16:07:03,042 readfish toml='docs/_static/human_chr_selection.toml'
2023-06-27 16:07:03,042 readfish.validate eJxtUNtOxCAUfOcrGp4NLU1N1MSnffcHmoZQOLRkuS0Xo38vdOOqWc8TM8wMc5gFNwYiS5CzdlsiWwnhc0HCO6W37rXD0nEW6UAmQhk8kZFNw7CGxHYuMOJSRkip6XQQL33fZxv6a0j/WAcjCWvZmPFHWIaUWQQulU47UReM0Gx5CL8bNFwb3Pv2YrljYm9aAyJr70jgqmXMEbYK07Igxy00y1VzyEfKxhEjq5u7uHPrS5HlHz9wQpnHDXI7z/jtxIY640AoxQ/dDVPyjBeUak0DzCvV3iluNV6ca3wxWd+x32J3dMo+1PUF6PdK3yz/3jlf17y0mxC9AJAHVT/nD9XmC4XQlII=
2023-06-27 16:07:03,047 readfish.validate Loaded TOML config without error
2023-06-27 16:07:03,047 readfish.validate Initialising Caller
2023-06-27 16:07:03,069 readfish.validate Caller initialised
2023-06-27 16:07:03,069 readfish.validate Initialising Aligner
2023-06-27 16:07:03,070 readfish.validate Aligner could not be initialised, see below for details
2023-06-27 16:07:03,071 readfish.validate Did not create or open an index
This command has a flag --no-check-plugins which will disable the loading and validation of plugins. However this is only recommended if you absolutely must not validate the plugins. For example the above TOML, missing the reference, would pass validation if using the --no-check-plugins flag.
The validate command also describes the experiment, and will error if contigs are listed in the targets that are not in the reference.
This can be disabled by passing the --no-describe flag.