Introducing ReadFish
Matt Loose 29 Nov 2020![]()
Today sees our preprint on adaptive sampling graduate to a full paper. This was a lot of work from a lot of people and we’re very happy to see the paper out.
Read Until to enrich >700 genes to mean 30x coverage on @nanopore single flowcell. Custom panels (up to 25,600 targets so far) as easy as providing a coordinate file. Read more at https://t.co/aZVOs23PKg - thanks to @alexomics, @Rorymatics and @DeepSeqNotts team. pic.twitter.com/0FBLlGQZdm
— Matt Loose (@mattloose) February 3, 2020
If you want to read the paper, in Nature Biotechnology, you can do so here.
We show in the final version that we can capture targets at depths in excess of 50x on a single MinION flowcell as can be seen in the figure below.
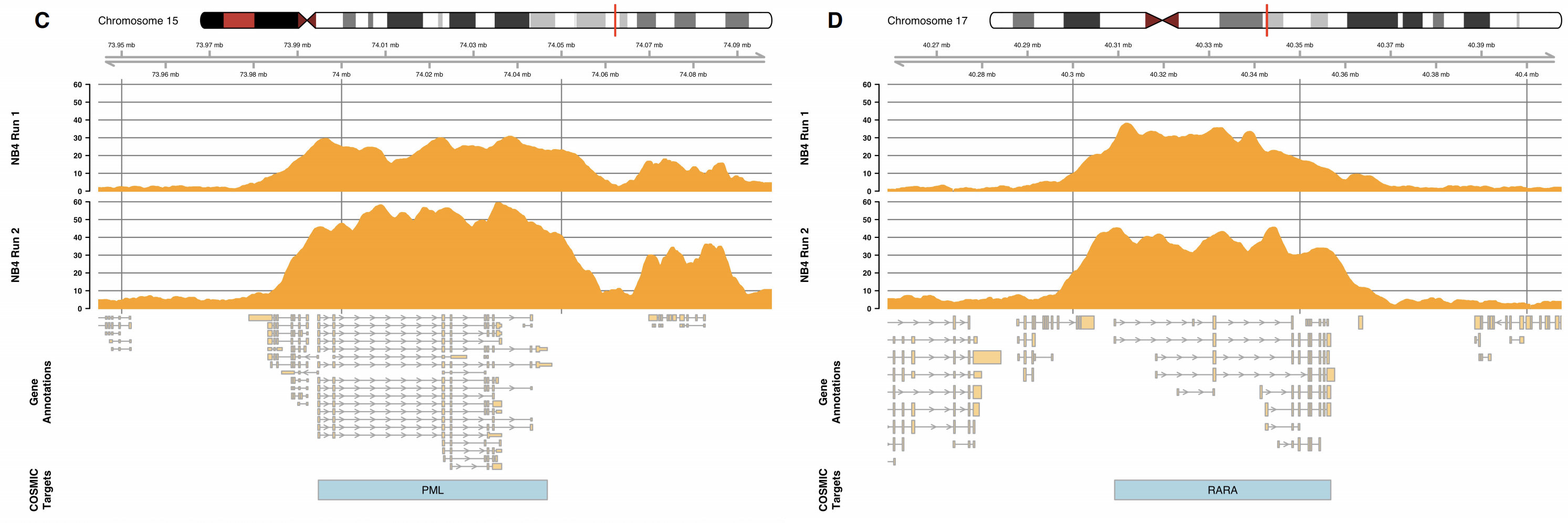
This blog is to talk a little about the changes we have made since the pre-print and to explain what we will be doing with our code in the next few months.
Firstly - why the name? Well - you gotta catch ‘em all! If you don’t know the link between Nanopore and fish then have a look at the software. We were asked to distinguish between our toolset and that provided by Oxford Nanopore - which makes sense given that with the recent release of MinKNOW for GridION Mk1, Oxford Nanopore have shipped an implementation for selecting individual regions from a genome or host enrichment or depletion. This may come to other platforms in the future.
So what does ReadFish do? Well - it enables you to select molecules from a library computationally - to catch whatever you like - on any platform with access to GPU basecalling (currently linux only). We built ReadFish as a platform to develop methods to exploit this unique ability of nanopore sequencers and the current toolkit is just the start.
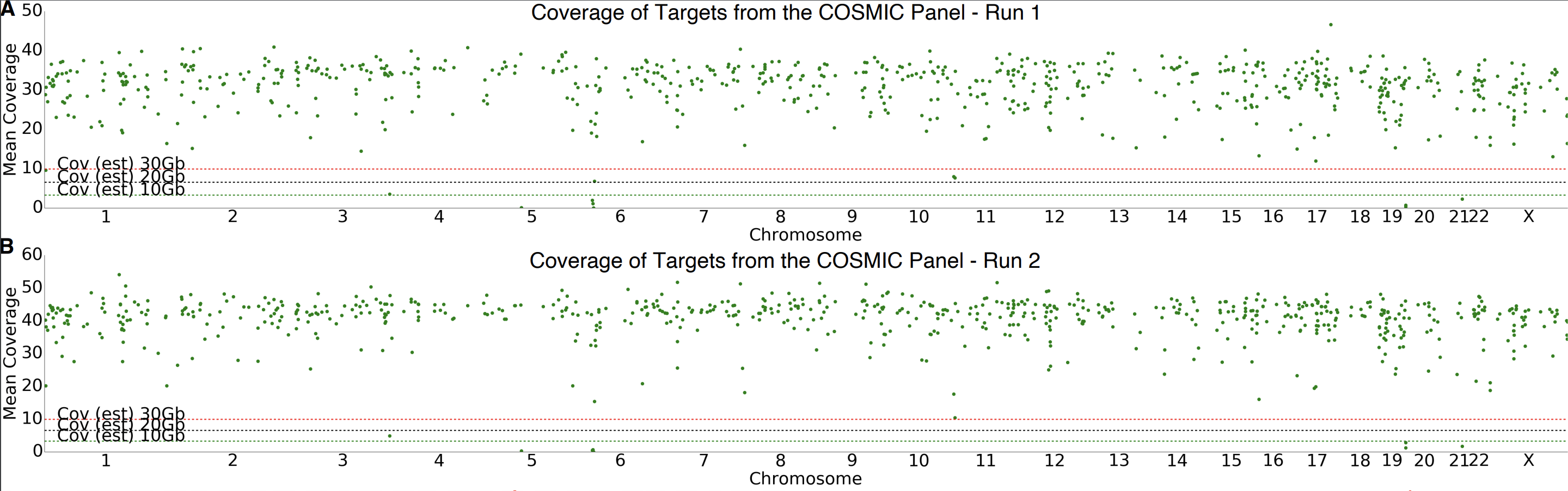
To date we have used ReadFish to capture many hundreds of genes to depths of around 30x (and sometimes up to 50x) on a single flowcell, enrich or deplete individual genomes from a background, and a personal favourite, identifying the composition of a sample and selecting individual components dynamically. ReadFish provides the ability to change what you are selecting for during a run in response to the data that has been seen to that point. This makes it adaptive.
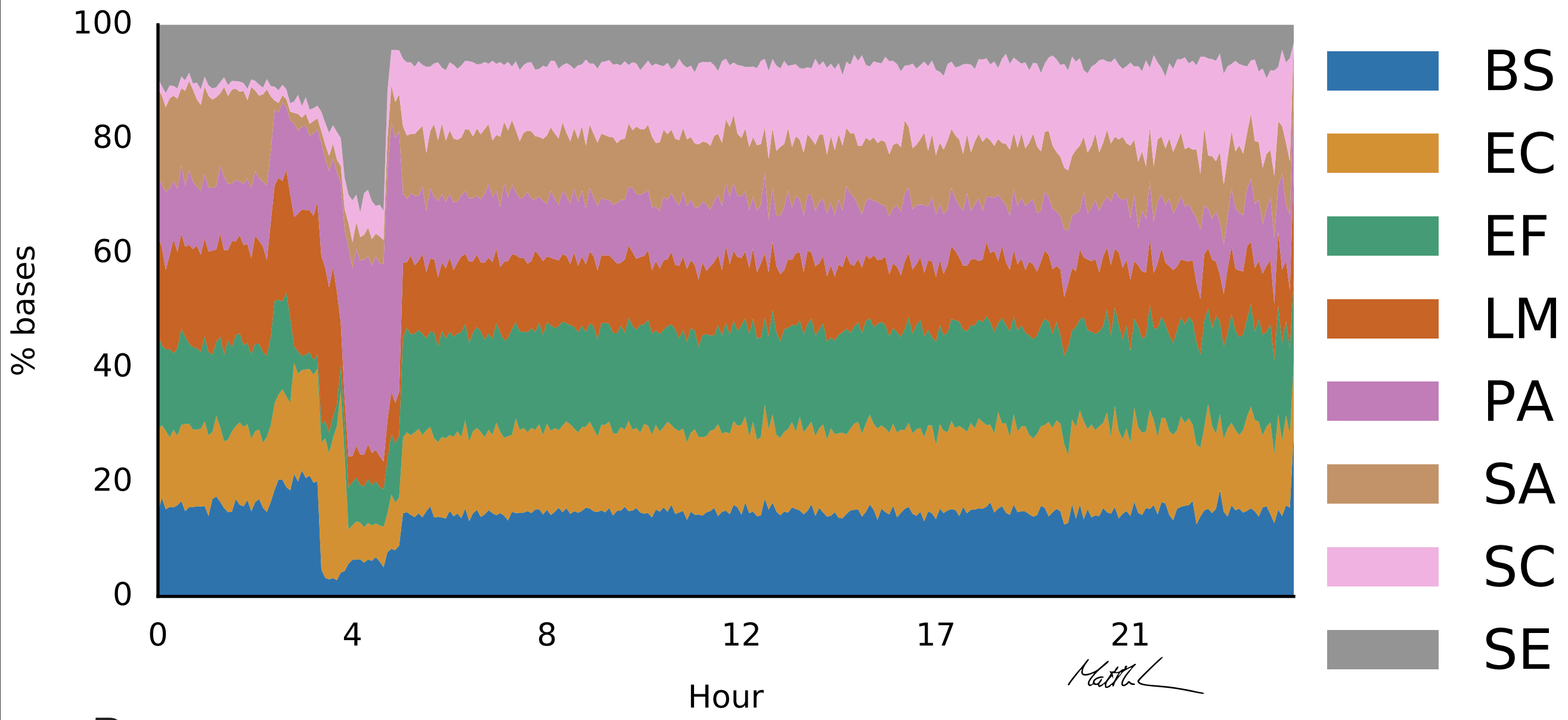
ReadFish currently enables adaptive sampling on any computer able to run GPU basecalling with Guppy. The computer and environment must be powerful enough to basecall at lesast two flowcells in real time and to handle mapping duties. For a human genome scale project, you need at least 16 Gb of RAM. ReadFish will be our vehicle for developing new methods around Read Until so keep an eye on this blog for updates.
Some of our future plans are described in a preprint from De Maio et al here.
Advice on how to run ReadFish can be found here.
Please pay particular attention to our instructions to test your experiment before running for real. Playback is your friend…